Afhankelijk van het soort WMO onderzoek gelden verschillende richtlijnen of regels. Zie hieronder de verschillende soorten:
- Onderzoek met geneesmiddelen (CTR/CTIS)
- Onderzoek met medische hulpmiddelen (MDR)
- Prestatiestudies naar In-Vitro Diagnostica (IVDR)
- Onderzoek met ioniserende straling
- Overig WMO-plichtig onderzoek
- WMO-plichtige zorgevaluatie (Veldnorm)
Onderzoek met geneesmiddelen (CTR/CTIS)
Clinical Trial Regulation (CTR)
Sinds 31 januari 2022 is de Clinical Trial Regulation, CTR) bekrachtigd. De European Clinical Trial Regulation (ECTR) is een Europese wet die de bestaande richtlijn Clinical Trials Directive (CTD; EU Directive 2001/20/EC) vervangt. Het doel van de ECTR is om de duur van de medisch-ethische toetsing van geneesmiddelenonderzoek gelijk te trekken voor alle landen binnen de EU en daarmee samenwerking tussen de lidstaten bij de ontwikkeling van geneesmiddelen makkelijker te maken.
Aanvraag CT-nummer binnen CTIS
Aanvraag CT-nummer binnen CTIS
De RvB van Amsterdam UMC heeft het Clinical Monitoring Center (CMC) aangewezen als sponsor administrator. Het CMC regelt voor u de aanvraag van een CT-nummer binnen CTIS. U vindt hier informatie over de nieuwe verordening (ECTR) en de rol van CTIS gedurende het gehele verloop van uw geneesmiddelenonderzoek.
Neem voor vragen contact op met CMC via ctis@amsterdamumc.nl
Voor een stappenplan met betrekking tot indiening kunt u ook terecht op de website van de CCMO die speciaal voor onderzoekers is opgezet: Geneesmiddelenonderzoek (CTIS).
Lokale uitvoerbaarheid CTIS studies
Lokale uitvoerbaarheid CTIS studies
Nadat deelname van Amsterdam UMC via CTIS is goedgekeurd, dient via Research Manager een aanvraag Lokale Uitvoerbaarheid (LU) te worden ingediend voor het verkrijgen van goedkeuring van de Raad van Bestuur Amsterdam UMC. Zie ook Lokale uitvoerbaarheid – Medisch Ethische Toetsingscommissie.
Onderzoek met medische hulpmiddelen (MDR)
Medical Devices Regulation (MDR)
Wanneer u wetenschappelijk onderzoek doet met een medisch hulpmiddel moet u voldoen aan verschillende wetten.
Per 26 mei 2021 is de Europese Verordening betreffende medische hulpmiddelen (MDR) van toepassing. Als gevolg daarvan zijn de regels voor de indiening, beoordeling en uitvoering van klinisch onderzoek met medische hulpmiddelen gewijzigd. Algemene informatie over de inwerkingtreding van de MDR vindt u op de website van de CCMO: Klinisch onderzoek naar medische hulpmiddelen (MDR).
Valt mijn onderzoek onder de MDR?
Valt mijn onderzoek onder de MDR?
Klinisch onderzoek naar de veiligheid, (klinische) prestaties en/of effectiviteit van het medisch hulpmiddel valt binnen de reikwijdte van de MDR. Ander onderzoek waarbij medische hulpmiddelen worden gebruikt in het kader van onderzoek (bijvoorbeeld het meten van een eindpunt), maar waarbij niet de veiligheid, prestaties en/of effectiviteit van het medisch hulpmiddel zelf worden onderzocht, valt buiten de reikwijdte van de MDR.
De EU-verordening medische hulpmiddelen (MDR) geeft de volgende definitie van klinisch onderzoek: “Systematisch onderzoek bij één of meer menselijke deelnemers dat wordt uitgevoerd om de veiligheid of de prestaties van een hulpmiddel te beoordelen”.
Wijze van indienen
Wijze van indienen
Alle onderzoeken moeten worden ingediend via het Onderzoeksportaal. De wijze waarop uw onderzoeksdossier vervolgens bij de toetsingscommissie terechtkomt, is afhankelijk van het wettelijke kader waar uw onderzoek onder valt. Hieronder staat ieder wettelijk kader uitgelegd.
- Klinisch onderzoek in het kader van conformiteitsdoeleinden (MDR artikel 62/74.2)
Bij klinisch onderzoek in het kader van conformiteitsdoeleinden (dat wil zeggen, in het kader van productontwikkeling en het verkrijgen/uitbreiden van een CE-markering) wordt het onderzoeksdossier eerst gevalideerd door de CCMO. Na een positieve validering draagt de CCMO uw dossier ter beoordeling over aan de toetsingscommissie (erkende METC of CCMO) die uw onderzoek zal beoordelen. De CCMO draagt uw dossier meestal over aan de toetsingscommissie van uw voorkeur.
- Post-market clinical follow-up investigations (MDR artikel 74.1) en overig klinisch onderzoek (MDR artikel 82)
Post-market clinical follow-up (PMCF) investigations (MDR artikel 74.1) en overig klinisch onderzoek (MDR artikel 82) hoeven niet gevalideerd te worden door de CCMO. Deze onderzoeken dient u in via het Onderzoeksportaal. De indiening gaat direct naar de Toetsingscommissie die u in het Onderzoeksportaal heeft gekozen.
Meer informatie vindt u hier.
Een overzicht van welke documenten u moet indienen voor welk wettelijk kader (artikel) vindt u hier.
Technisch advies voor gebruik van medisch apparaten of medische software in Amsterdam UMC
Technisch advies voor gebruik van medisch apparaten of medische software in Amsterdam UMC
Volgens eerder vastgesteld beleid werd bij de inzet van niet-CE-gemarkeerde medische hulpmiddelen of CE-gemarkeerde medische hulpmiddelen die buiten beoogd gebruik worden toegepast, gevraagd om een technisch advies bij de METC-indiening mee te sturen. Vanwege de overlap van dit technisch inhoudelijk advies met de METC beoordeling zelf, is besloten dat het technisch advies is komen te vervallen. Een aanvraag via het loket AdviesMedischeHulpmiddelen@amsterdamumc.nl is dus niet langer nodig.
Wel is, in overeenstemming met het Amsterdam UMC aanschafbeleid (link is enkel door Amsterdam UMC-ers te openen), een beoordeling door de TMTa (Toetsingscommissie Medische Technologie apparatuur) nodig voor ieder medisch apparaat of medische software dat u in het kader van klinisch wetenschappelijk onderzoek introduceert in Amsterdam UMC. De TMTa beoordeelt de inzetbaarheid en de mogelijkheid om de medische apparatuur of software te beheren. Dit is verplicht voordat u het hulpmiddel kunt inzetten in Amsterdam UMC. Deze beoordeling vraagt u aan door het aanschafdossier in te vullen, de grote groene “INVULLEN” knop te selecteren op de startpagina van K2 (link is enkel door Amsterdam UMC-ers te openen) en vervolgens te kiezen voor “Aanschafdossier Medische/Lab apparatuur of software”. Let wel, het is van toepassing voor ieder medisch apparaat of medische software: dus of het hulpmiddel nu wel of geen CE-markering heeft, wel of niet binnen beoogd gebruik wordt toegepast, wordt geschonken, geleend of aangeschaft, in elke situatie is het aanschafdossier nodig.
Belangrijk om te weten is dat de beoordeling van de TMTa niet mee hoeft te worden gestuurd met de METC-aanvraag. De twee trajecten kunnen dus parallel lopen, zodat de vertraging zo klein mogelijk is. Het is wel zeer aan te raden om het aanschafdossier in te vullen voordat u de METC-aanvraag indient, zodat u niet voor de verassing komt te staan dat uw studie is goedgekeurd door de METC, maar het hulpmiddel nog niet ingezet mag worden.
Voor vragen of voor hulp bij het invullen van het aanschafdossier, kunt u terecht bij het loket AdviesMedischeHulpmiddelen@amsterdamumc.nl.
Zelf een IMDD samenstellen
Zelf een IMDD samenstellen
Voor klinisch wetenschappelijk onderzoek met medische hulpmiddelen die zonder CE-markering of buiten beoogd gebruik worden ingezet, heeft u een Investigational Medical Device Dossier (IMDD) nodig voor uw METC-indiening.
Er zijn documenten ontwikkeld om eenvoudiger een IMDD (of IMDD-light) samen te stellen. Zo is er een handleiding voor het schrijven van een IMDD is beschikbaar op K2, evenals een voorbeeld IMDD met bijbehorende risicoanalyse. Voor laag-risico hulpmiddelen kan soms een IMDD-light voldoende zijn, dit staat beschreven in hoofdstuk 3 van de IMDD handleiding. Sinds kort is ook een voorbeeld IMDD-light beschikbaar op K2 zodat onderzoekers daar hun voordeel mee kunnen doen. Deze documenten zijn hier te vinden:
- Klik hier voor een handleiding (link is enkel door Amsterdam UMC-ers te openen)
- Klik hier voor een voorbeeld IMDD (link is enkel door Amsterdam UMC-ers te openen)
- Klik hier voor de risicoanalyse (link is enkel door Amsterdam UMC-ers te openen)
- Klik hier voor een voorbeeld IMDD-light (link is enkel door Amsterdam UMC-ers te openen)
Voor vragen over bovenstaande documenten verwijzen wij u graag door naar adviesmedischehulpmiddelen@amsterdamumc.nl.
Prestatiestudies naar In-Vitro Diagnostica (IVDR)
In-Vitro Diagnostics Regulation (IVDR)
Specifieke regels voor de indiening, beoordeling en uitvoering van prestatiestudies naar medische hulpmiddelen voor in-vitro diagnostica (IVD’s) zijn vastgelegd in de EU-verordening 2017/746, ook bekend als de In-Vitro Diagnostics Regulation (IVDR). IVD’s moeten sinds 26 mei 2022 voldoen aan nieuwe Europese regels.
Wat is een IVD?
Wat is een IVD?
De IVDR geeft de volgende definitie van een medisch hulpmiddel voor in-vitrodiagnostiek (IVD):
“Elk medisch hulpmiddel dat een reagens, een reactief product, een kalibrator, een controlemateriaal, een kit, een instrument, een apparaat, een toestel, software of een systeem is dat afzonderlijk of in combinatie wordt gebruikt, en door de fabrikant is bestemd om te worden gebruikt voor het in-vitro-onderzoek van specimens die afkomstig zijn van het menselijk lichaam, met inbegrip van donorbloed en -weefsel, uitsluitend of hoofdzakelijk met het doel informatie te verschaffen over een of meer van de volgende elementen:
- over een fysiologisch of pathologisch proces of een fysiologische of pathologische toestand;
- over aangeboren lichamelijke of geestelijke beperkingen;
- over de predispositie voor een medische aandoening of een ziekte;
- om de veiligheid en de mate van verenigbaarheid met potentiële ontvangers te bepalen;
- om de respons of de reacties op de behandeling te voorspellen;
- om therapeutische maatregelen te bepalen of te monitoren.”
Recipiënten voor specimens worden ook als medisch hulpmiddel voor in-vitrodiagnostiek beschouwd.
Definitie prestatiestudies
Definitie prestatiestudies
Onderzoek naar de prestatie van een IVD valt binnen de reikwijdte van de IVDR. Andere studies waarbij IVD’s worden gebruikt in het kader van onderzoek (bijvoorbeeld het meten van een eindpunt), maar waarbij niet de prestatie van het IVD zelf wordt onderzocht, vallen buiten de reikwijdte van hoofdstuk VI van de IVDR. Deze studies vallen mogelijk wel onder de reikwijdte van de Wet medisch-wetenschappelijk onderzoek met mensen (WMO) of de Embryowet.
De IVDR geeft de volgende definitie van een prestatiestudie: “een studie die wordt uitgevoerd ter bepaling of bevestiging van de analytische of klinische prestaties van een hulpmiddel”. Met analytische prestaties wordt het vermogen bedoeld van een IVD om een specifiek analyt correct te detecteren of te meten. Met klinische prestatie wordt het vermogen bedoeld om resultaten te leveren die klinische relevant zijn.
Kaders prestatiestudies
Kaders prestatiestudies
De IVDR kent de volgende kaders voor prestatiestudies, met verschillende vereisten en procedures:
- Prestatiestudies met companion diagnostics (CDx) (IVDR artikel 58, lid 2)
- waarbij de chirurgisch invasieve bemonstering wordt uitgevoerd uitsluitend ten behoeve van de prestatiestudie;
- die een interventionele klinische prestatiestudie betreft waarbij de testresultaten van invloed kunnen zijn op besluiten inzake patiëntenzorg en/of kunnen worden gebruikt als richtsnoer voor de behandeling, of
- waarbij de uitvoering van de studie bijkomende invasieve procedures of andere risico’s voor de deelnemers van de studies omvat.
- Post-market performance follow-up (PMPF) met extra invasieve en/of belastende procedures (IVDR artikel 70, lid 1)
- Prestatiestudies zonder risico voor onderzoeksdeelnemer (IVDR artikel 57). Deze hoeven niet getoetst te worden door een erkende METC/CCMO.
- Prestatiestudies met risico voor onderzoeksdeelnemer (IVDR artikel 58, lid 1 en artikel 70, lid 2)
Met onderstaande flowchart kunt u bepalen onder welk kader uw prestatiestudie valt:
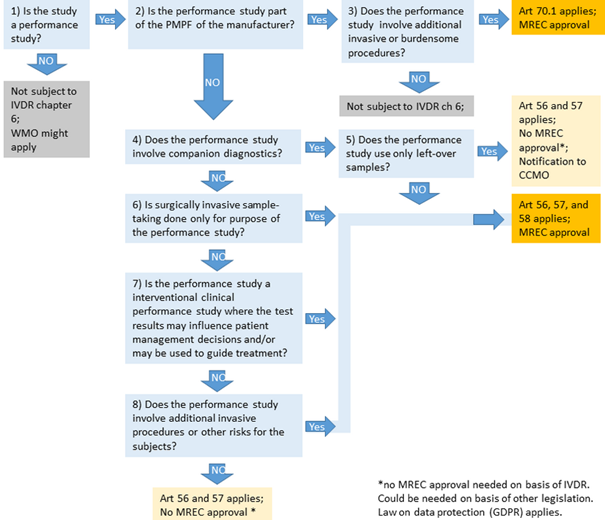
Wijze van indienen
Wijze van indienen
In de IVDR en/of de WMO is vastgesteld dat bepaalde IVD-prestatiestudies vooraf door een bevoegde toetsingscommissie moeten worden beoordeeld. Daarnaast moeten sommige onderzoeksdossiers eerst worden gevalideerd door de CCMO alvorens deze beoordeeld kunnen worden:
- IVDR artikel 58 en 70.2 (prestatiestudies met risico voor onderzoeksdeelnemer) – validatie door de CCMO
- Na een positieve validatie draagt de CCMO uw dossier ter beoordeling over aan de toetsingscommissie (erkende METC of CCMO). De CCMO houdt bij de toewijzing van uw dossier aan de erkende METC rekening met uw METC van voorkeur die u kenbaar kunt maken in de aanbiedingsbrief bij het dossier.
- In ToetsingOnline kunt u voor uw dossier alleen de CCMO selecteren als toetsingscommissie. Na de validering past de CCMO de toetsingscommissie aan in ToetsingOnline.
- Voor meer informatie zie: Validering van IVD-prestatiestudies door de CCMO
- IVDR artikel 70.1 (Post-market performance follow-up (PMPF) met extra invasieve en/of belastende procedures) – direct indienen bij een erkende METC
Voor meer informatie over de wijze van indienen (verschillende kaders), zie: Wijze van indiening
Voor een overzicht van in te dienen documenten voor IVD-prestatiestudies, zie: Overzicht in te dienen documenten voor IVD-prestatiestudies.
Onderzoek met ioniserende straling
Bij gebruik van ioniserende straling ten behoeve van het onderzoek moet de dosis voor de deelnemer van de studie vooraf worden bepaald. Voor conventionele röntgenonderzoeken kan u gebruik maken van de dosis in onderstaande tabel.
Mocht uw toepassing van ioniserende straling niet voorkomen in onderstaande tabel dan kunt u een verzoek voor een stralingsberekening indienen door het aanvraagformulier stralingsberekening in te vullen en te mailen naar de betreffende klinisch fysicus (email in het formulier).
Overig WMO-plichtig onderzoek
Een overige medisch wetenschappelijke studie valt niet onder de CTR, MDR en/of IVDR, maar is wel WMO-plichtig.
Onderzoek valt onder de reikwijdte van de WMO als voldaan is aan de twee volgende voorwaarden:
- het is medisch-wetenschappelijk onderzoek en
- de onderzoeksdeelnemers worden onderworpen aan handelingen en/of krijgen een gedragswijze opgelegd, zoals bedoeld in de definitie van medisch-wetenschappelijk onderzoek in artikel 1 lid 1 sub b van de WMO.
Ad 1. Medisch wetenschappelijk onderzoek is onderzoek dat als doel heeft het beantwoorden van een vraag op het gebied van ziekte en gezondheid (etiologie, pathogenese, verschijnselen/symptomen, diagnose, preventie, uitkomst of behandeling van ziekte), door het op systematische wijze vergaren en bestuderen van gegevens. Het onderzoek beoogt bij te dragen aan medische kennis die ook geldend is voor populaties buiten de directe onderzoekspopulatie.
Ad 2. Er is sprake van een gedragswijze of een handeling, indien de handeling inbreuk maakt op de lichamelijke of psychische integriteit van de onderzoeksdeelnemers.
WMO-plichtige zorgevaluatie
Verklaring bestaande zorg
Alleen van toepassing bij evaluatie van bestaande zorg, om in aanmerking te komen voor toetsing volgens de Veldnorm.